(c) Elliot Israel, MD, Assistant Professor of Medicine, Harvard Medical School.
IntroductionFive to thirty percent of asthmatics may be sensitive to aspirin (ASA). The first case of ASA sensitivity was described in 1902 and, in 1968, Samter and Beers described a triad consisting of asthma, aspirin sensitivity, and nasal polyps. Aspirin-induced asthma (AIA) was originally thought to be due to an "allergy" to aspirin; however, we now know that most patients also react to structurally distinct nonsteroidal antiinflammatory drugs (NSAIDs), suggesting that nonallergic mechanisms are operative in this syndrome. Clinical PresentationAIA tends to present in the third to fourth decade in individuals not previously sensitive to aspirin or NSAIDs. Reactions are usually slow in onset, thirty minutes to two hours after ingestion, and may be slow to resolve. In addition to wheezing, reactions are usually accompanied by profound nasal symptoms. Facial flushing, angioedema, and gastrointestinal symptoms can also occur. Hives are uncommon and occur in a distinct syndrome characterized by aspirin sensitivity without asthma, called ASA-induced urticaria/angioedema. Patients with AIA tend to have more severe asthma than patients without aspirin sensitivity. However, complaints related to nasal polyposis and chronic rhinosinusitis may actually be more troubling to the patient on a day-to-day basis than recurrent bronchospasm. PathophysiologyPatients with AIA react to structurally distinct
compounds that have in common their ability to inhibit the constitutively
expressed form of the cyclooxygenase enxyme — cyclooxygenase-1.
Cyclooxygenase-1 constitutively catalyzes the formation of lipid mediators
such as prostaglandins and thromboxanes from cell membrane arachidonic
acid. However, arachidonic acid can undergo an alternate pathway of
metabolism involving the enzyme 5-lipoxygenase. | |

| |
|
The 5-lipoxygenase products of arachidonic acid, which
include the cysteinyl leukotrienes, LTC4, LTD4, and LTE4, are potent
inflammatory mediators; they can act as bronchoconstrictors, induce mucous
secretion, promote airway edema, and can attract eosinophils into the
airways. Other products of this pathway are also known to be potent chemotactic
agents and mucous secretagogues.
While the precise mechanism leading to increased leukotriene production in AIA
patients is unclear, overexpression of the enzyme LTC4 synthase appears to
be associated with this abnormality. In fact, preliminary data suggest that
patients with a particular polymorphism of the LTC4 synthase gene may be
greater than three times more likely to have AIA than those without the polymorphism.
Additionally, the cyclooxygenase product PGE2 may play a role in down-regulating
leukotriene production, and it is possible that inhibition of its synthesis, by
blockade of cyclooxygenase, may play a role in increased leukotriene production.
DiagnosisWhile the prevalence of AIA varies with the population
studied, it has been reported in up to 19 percent of steroid-dependent
asthmatics. It is especially common in
asthmatics with nasal polyps and sinusitis, possibly approaching a
prevalence of 20 to 30 percent in these subjects. It is likely that a
prevalence of five to 10 percent is a reasonable estimate among asthmatics
with moderate or severe disease.
| |
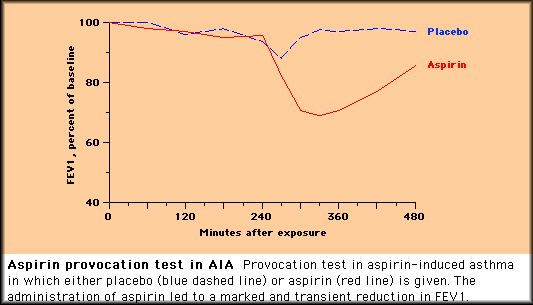
| |
|
Unless urinary leukotrienes are measured, the use of a placebo control is necessary because many patients will have a nonspecific spontaneous decline in pulmonary function over the several hours required for ASA challenge. Non-AIA patients do not overproduce leukotrienes in response to cyclooxygenase blockade, and thus a rise in urinary leukotrienes is specific for AIA. Due to potentially serious reactions, such tests should be done in a hospital setting. Lysine-ASA, a soluble form of aspirin not available in the United States, has been used by inhalation for bronchoprovocation challenge in the outpatient setting outside the United States. TreatmentThe usual guidelines for asthma therapy also apply to the NSAID-intolerant asthmatic. Standard therapy utilizing a step-care approach, such as that described in the "Guidelines for the Diagnosis and Treatment of Asthma" published by the NHLBI, is appropriate NSAID AvoidanceNSAID avoidance is crucial in the care of
asthmatics with aspirin sensitivity. AIA can be induced by aspirin and the
commonly available NSAIDs. | |

| |
|
Acetaminophen is a generally safe substitute. It does, however, have weak cyclooxygenase inhibitory properties, and very rarely patients who are very sensitive to ASA may have an adverse reaction to acetaminophen if taken at high doses. Choline magnesium salicylate and salicylic acid have weak to absent cyclooxygenase inhibitory properties and can also be used with appropriate caution in these patients. DesensitizationAIA patients can be desensitized to ASA or NSAIDs
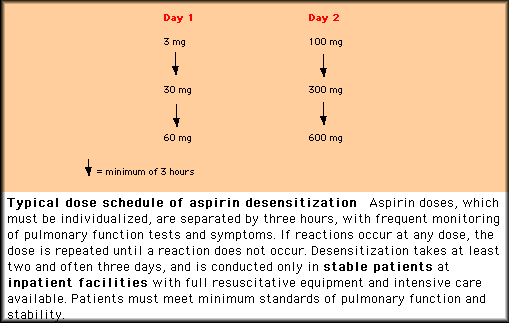
using a protocol that requires one to three days of inpatient treatment
and continuous daily aspirin ingestion to maintain the desensitized state. | |
| |
|
Such treatment may be desirable in clinical settings requiring continuous NSAID ingestion such as a rheumatologic condition or inhibition of platelet aggregation for coronary artery disease prophylaxis. Aspirin desensitization has been reported to improve the upper and lower airway disease of asthmatics with NSAID intolerance and is utilized in some centers for treatment of refractory AIA patients. Sinus DiseaseSinus and nasal disease should be actively evaluated and treated in patients with AIA. Nasal steroids should be used aggressively to retard polyp growth and to promote regression. While nasal polypectomy may improve the upper airway symptoms and reduce the occurrence of sinus infections, the benefits of this procedure may be short-lived since the polyps almost always recur. Sinus infections in ASA-sensitive patients frequently require prolonged (six to eight week) courses of antibiotic therapy for adequate resolution. Sinus surgery to improve drainage may be necessary and should be considered on a case by case basis. Leukotriene ModifiersThe identification of the critical role of
leukotrienes in AIA has led to the possibility of more specific and
effective therapy. Inhibitors of leukotriene synthesis, as well as
leukotriene receptor antagonists, have been reported to blunt the response
to aspirin challenge. In addition,
pretreatment with a 5 lipoxygenase inhibitor has been shown to ablate the
effects of ASA ingestion in ASA-sensitive asthmatics.
Both zileuton (a 5-lipoxygenase inhibitor and thus an inhibitor of leukotriene
synthesis) and zafirlukast and montelukast (LTD4 receptor antagonists) are
currently available in the United States. These agents have been shown to
improve pulmonary function and decrease ß-agonist use in AIA patients, even
in those already being treated with corticosteroids. In view
of our current knowledge, these drugs should be the cornerstone of
treatment for the AIA patient. | |
References:
| |
 |
 |